31/08/2023
Ioner i opløsning er fundamentale for en lang række fysiske, kemiske og biologiske processer. Fra nerveimpulser i kroppen til elektrokemiske reaktioner i batterier og industrielle processer, spiller ioners evne til at bevæge sig frit en afgørende rolle. Men hvordan bevæger disse mikroskopiske ladede partikler sig egentlig gennem et solvent som vand? Og hvilke faktorer dikterer deres hastighed og retning? I årtier har forskningen forsøgt at afdække de komplekse interaktioner mellem ioner og deres omgivelser, især solventmolekylerne. Denne artikel vil udforske forholdet mellem ioners solvationsdynamik og deres mobilitet, og vi vil se, hvordan moderne teorier og simuleringsmetoder har revolutioneret vores forståelse, især når det kommer til de mere komplekse polyatomiske ioner.
Hvad er Ionmobilitet?
Ionmobilitet beskriver, hvor hurtigt en ion bevæger sig gennem en opløsning under påvirkning af et elektrisk felt. Når et elektrisk felt (E) påføres, vil ioner accelerere, indtil friktionskræfterne i opløsningen præcist modvirker accelerationskraften. På dette tidspunkt opnår ionen en stabil drivhastighed (s). Forholdet mellem drivhastigheden og feltstyrken definerer ionmobiliteten (u):
u = s/E
Ifølge Stokes' lov for en sfærisk partikel med radius Reff i et medium med viskositet η, er friktionskraften (f) givet ved:
f = 6πηReffs
Da den elektriske kraft på ionen er f = zeE (hvor z er ladningen på partiklen og e er elementarladningen), kan vi sætte disse kræfter lig med hinanden for at finde et alternativt udtryk for mobiliteten:
zeE = 6πηReffs ⇒ s = zeE / (6πηReff)
Hvilket fører til:
u = ze / (6πηReff)
Dette viser, at mobiliteten øges med ionens ladning, men mindskes med stigende viskositet og effektiv størrelse.
Anomale Mobiliteter
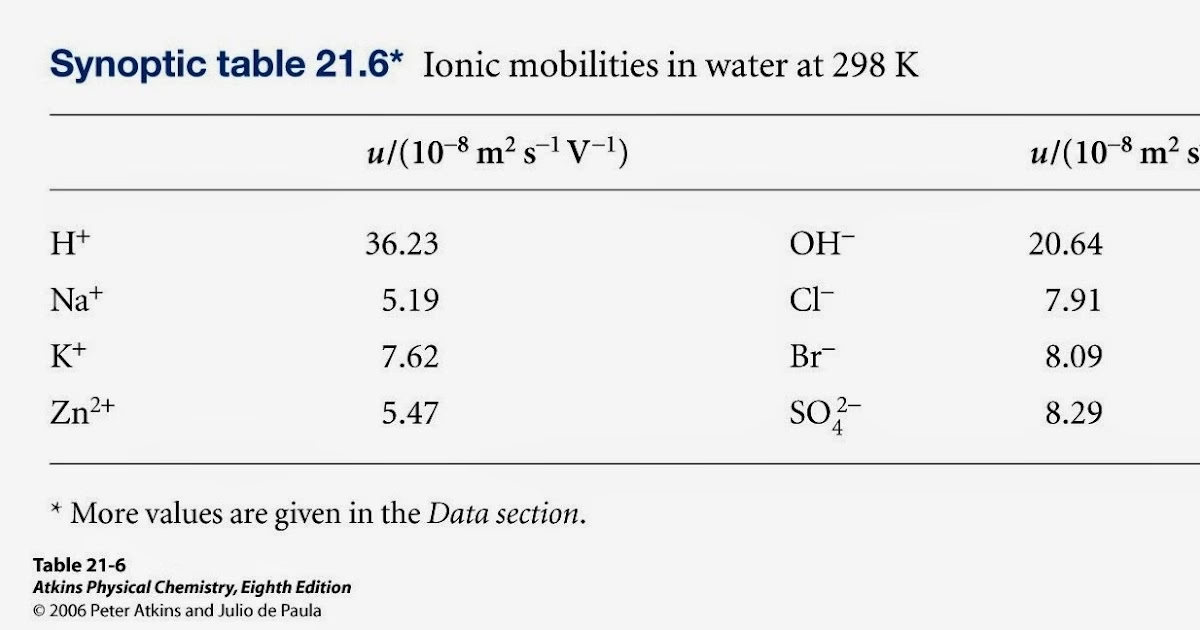
Et hurtigt blik på mobiliteterne for nogle almindelige ioner i vand ved 298 K afslører bemærkelsesværdige afvigelser, især for protoner (H+) og hydroxidioner (OH-). Deres mobiliteter er markant højere end forventet sammenlignet med andre ioner af lignende størrelse. Dette fænomen forklares af Grotthus-mekanismen.
I stedet for at disse ioner diffunderer fysisk gennem opløsningen, overfører de simpelthen en hydrogenbinding til deres nærmeste nabo i den korrekte retning, mens naboen samtidigt gør det samme. Dette betyder, at den effektive ladning kan overføres gennem en lang kæde af vandmolekyler, hvor hver proton kun bevæger sig en kort afstand, men processen sker samtidigt. Det faktum, at mobiliteten af H+ og OH- er 5-10 gange højere end for andre små ladede ioner, antyder, at længden af overførselskæderne er i størrelsesordenen 5-10 molekyler.
Her er en oversigt over mobiliteter for nogle almindelige ioner i vand ved 298 K:
| Kation | Mobilitet (10-8 m2s-1V-1) | Anion | Mobilitet (10-8 m2s-1V-1) |
|---|---|---|---|
| H+ | 36.23 | OH- | 20.64 |
| Li+ | 4.01 | F- | 5.74 |
| Na+ | 5.19 | Cl- | 7.92 |
| K+ | 7.62 | Br- | 8.09 |
| Rb+ | 8.06 | I- | 7.96 |
| Cs+ | 8.00 | CO32- | 7.46 |
| Mg2+ | 5.50 | NO3- | 7.41 |
| Ca2+ | 6.17 | SO42- | 8.29 |
Ionmobilitet vs. Diffusivitet
Inden for studiet af massetransport i elektrolytopløsninger er det vigtigt at skelne mellem forskellige former for ionbevægelse. Mens ionmobilitet primært handler om ioners bevægelse under påvirkning af et elektrisk potentialegradient (migration), beskriver diffusivitet (D) ioners bevægelse drevet af koncentrationsgradienter. Nernst-Planck-teorien er en central model, der integrerer disse bevægelsestyper sammen med konvektion (bevægelse med bulkvæskens hastighed).
Selvom de er forskellige koncepter, er ionmobilitet og diffusivitet tæt forbundet gennem Nernst-Einsteins relation, som ofte bruges til at udlede den ene fra den anden, især ved uendelig fortynding. Begge er afgørende for at forstå, hvordan ioner fordeler sig og reagerer i et givet miljø.
Sammenbruddet af Kontinuummodeller
I årtier har forskere anvendt hydrodynamiske kontinuummodeller, såsom Stokes-Einstein-relationen, til at beskrive ioners bevægelse i opløsninger. Disse modeller antager, at solventet kan behandles som et viskøst, kontinuerligt medium, og at solutpartiklen (ionen) er meget større end solventmolekylerne. Imidlertid har eksperimentelle observationer af ionmobilitet, især for små, stive monoatomiske ioner som Li+, Na+ og K+, samt halogenidioner, vist et spektakulært sammenbrud af disse modeller. Dette fænomen er populært kendt som "Walden-produktets sammenbrud", hvor produktet af den begrænsende ioniske ledningsevne (Λ0) og solventets viskositet (η0) ikke udviser den forventede konstante adfærd.
Årsagerne til kontinuummodellernes fiasko er mangefoldige:
- Ignorering af molekylær detalje: De overser den molekylære natur af ladnings-solvent-interaktioner og undervurderer derved solut-solvent- og solvent-solvent-korrelationer.
- Utilstrækkelig beskrivelse af solventdynamik: Teorierne blev udviklet årtier før opdagelsen af ultrahurtig solvation dynamik i vand og andre polære solventer.
- Lange rækkevidde og anisotropi: Interaktionen mellem ionens ladning og vandmolekylernes dipolmoment er langtrækkende og anisotrop.
- Sammenlignelig størrelse: Ionens størrelse er ofte ikke meget større end vandmolekylerne selv, hvilket gør antagelsen om et "kontinuum" ugyldig.
En central faktor i dette sammenbrud er fænomenet dielektrisk friktion. Dette er en ekstra friktion, ud over Stokes' friktion, der opstår på grund af interaktionen mellem ionens ladning og solventets dipoler. Kontinuummodellerne har tendens til at overvurdere denne dielektriske friktion for små ioner og kan ikke korrekt redegøre for størrelsesafhængigheden på tværs af kationserien. Dette er især tydeligt for de mindste kationer.
For at opnå en dybere forståelse af ioners bevægelse er det derfor nødvendigt at ty til mere sofistikerede mikroskopiske tilgange, der kan indfange disse molekylære interaktioner og dynamikker.
Monoatomiske Ioner: Friktion og Solvation
Forskningen har længe fokuseret på de enklere, stive monoatomiske ioner som Li+, Na+, K+, Cl- og Br- på grund af deres fundamentale betydning i fysisk kemi. Selvom disse ioner mangler de rotationelle frihedsgrader, der findes i polyatomiske ioner, udviser deres translationelle diffusion en bemærkelsesværdig ikke-monoton afhængighed af den inverse ionstørrelse – et fænomen, der er centralt i Walden-produktets sammenbrud.
Ved at analysere friktionskræfterne på disse ioner kan vi skelne mellem to hovedkomponenter: Stokes' friktion (ζSE) og dielektrisk friktion (ζDF). Stokes' friktion stiger som forventet med ionens størrelse (fra Li+ til Cs+), da større partikler oplever mere modstand fra solventet. Derimod viser den dielektriske friktion et skarpt fald efter Na+-ionen. Dette skyldes den komplekse interaktion mellem ionens ladning og vandmolekylernes dipoler. Interessant nok krydser de to friktionslinjer hinanden ved kalium (K+), hvilket indikerer, at denne ion oplever lignende Stokes' og dielektrisk friktion.
En afgørende indsigt i forståelsen af dielektrisk friktion kommer fra opdagelsen af ultrahurtig solvation dynamik i vand. Studier har vist, at en betydelig del (60-70%) af energirelakseringen omkring en ion i vand sker inden for de første 100 femtosekunder. Denne ultrahurtige proces har en signifikant rolle i at reducere den samlede dielektriske friktion på en bevægende ion. Fordi den indledende del af kraft-kraft-tidskorrelationsfunktionen forfalder med en ultrahurtig hastighed, mindskes bidraget til den totale friktion. Hvis de ultrahurtige komponenter fjernes progressivt, nærmer den begrænsende ioniske ledningsevne sig forudsigelsen fra Zwanzigs teori, hvilket understreger betydningen af denne hurtige dynamik.
Mikroskopiske Modeller og Simuleringsindsigter
For at overvinde begrænsningerne ved kontinuummodeller er der udviklet mikroskopiske teorier, især Modekoblingsteori (MCT). MCT er en molekylær tilgang, der inkluderer rumlige og orienteringsmæssige korrelationer samt dynamik på molekylære længdeskalaer. Den tager også højde for lokale strukturelle ændringer i solventet på grund af ionernes tilstedeværelse. Gennem en selvkonsekvent tilgang, der forbinder solut- og solventdynamik, har MCT vist sig at stemme godt overens med eksperimentelle resultater for monoatomiske ioner, f.eks. i methanol.
Computersimuleringer har yderligere understøttet disse teoretiske fremskridt. De giver en detaljeret forståelse af, hvordan ioner interagerer med vandmolekyler på et molekylært niveau. For eksempel viser sandsynlighedsfordelingen af kvadratisk forskydning for alkali-ioner, at mindre ioner som Li+ og Na+ opfører sig ens, mens større ioner som K+ og Rb+ opfører sig markant anderledes. Dette skyldes, at mindre ioner, trods deres potentielt hurtige bevægelse, danner et stærkt elektrisk felt, der skaber en "solvent-berg" struktur omkring dem, hvilket MCT kan fange gennem solut-solvent-struktur og selvkonsekvens.
Endelig er det vigtigt at bemærke, at vandmolekylernes polariserbarhed også spiller en signifikant rolle. Simuleringer med polariserbare vandmodeller har vist, at hydrogenbindingsafslapningstider forlænges, og aktiveringsenergien for at bryde hydrogenbindinger øges. Dette indikerer, at nøjagtige simuleringsmodeller skal inkludere polariserbarhed og/eller ladningsoverførsel for at opnå god overensstemmelse med eksperimentelle data, især i studiet af ioners hydreringsskaller og deres dynamik.
Polyatomiske Ioner: Et Nyt Kompleksitetsniveau
Mens meget opmærksomhed historisk har været rettet mod monoatomiske ioner, er polyatomiske ioner af enorm betydning i biologiske og kemiske processer – tænk på nitrat, acetat, sulfat og ammonium. Men deres dynamik er langt mere kompleks, og først for nylig er de "anomalier", de udviser, blevet fuldt ud anerkendt.
I modsætning til stive, sfæriske monoatomiske ioner, udviser mellemstore til store polyatomiske ioner (såsom nitrat og acetat) en slående forskel i diffusivitet, selv når de har lignende ionradier. For eksempel kan nitrationer (NO3-) have en markant højere diffusivitet (75-100%) end ioner som iodat (IO3-) eller acetat (CH3COO-), selvom de har sammenlignelige størrelser. Ifølge Stokes-Einstein-relationen burde de udvise ens diffusivitet, men det gør de ikke.
Oprindelsen til denne kompleksitet er mangesidig:
- Fordelte ladninger og geometri: Friktionen på en polyatomisk ion afhænger ikke kun af vandmolekylernes translationelle og rotationelle bevægelser, men også af ionens egne rotationelle og translationelle bevægelser. Fordelte ladninger på tværs af heteroatomer og ionens specifikke geometri spiller en afgørende rolle.
- Kobling af bevægelser: Bevægelserne af polyatomiske ioner er indviklet forbundet med vandmolekylernes springbevægelser i væskefasen. Derudover er ionens egen rotationelle bevægelse koblet til dens translationelle bevægelse – et fænomen kendt som translation-rotation kobling. Dette bidrager signifikant til den samlede dynamik.
Eksempler på Polyatomiske Ioners Dynamik
Diatomiske ioner: Cyanid (CN-) vs. Kulilte (CO)
Selv for relativt simple diatomiske arter er en pålidelig kvantitativ beskrivelse af translationel og rotationel diffusion udfordrende. Sammenligning af cyanidionen (CN-) og kuliltemolekylet (CO) i vand giver interessante indsigter. CO-molekylet, som er uladet, udviser hurtigere translationel og rotationel dynamik end cyanidionen. Selvom CO roterer hurtigere, er den mindre forbundet med vanddynamikken. Cyanidionen, derimod, er bedre forbundet på grund af tilstedeværelsen af ladninger og drager fordel af vandmolekylernes springdynamik, hvilket afspejles i deres bevægelsesmønstre. Dette understreger, hvordan ladningsfordelingen påvirker koblingen til solventet.
Nitrat (NO3-), Acetat (CH3COO-) og Sulfat (SO42-)
Disse ioner repræsenterer forskellige grader af kompleksitet:
- Nitrationen (NO3-): Med sin plane geometri og symmetriske ladningsfordeling roterer nitrationen markant hurtigere end acetat. Dens rotationelle bevægelse er stærkt koblet til vandmolekylernes orienteringsspring, hvilket betyder, at når ion-vand-hydrogenbindinger brydes, udviser både ionen og vandet store rotationelle springbevægelser, der er korrelerede. Denne kobling bidrager til dens relativt høje diffusivitet.
- Acetat-ionen (CH3COO-): Denne ion har en asymmetrisk ladningsfordeling. De partielle ladninger på dens atomer varierer, hvilket får vanddensiteten til at forskydes mod de to iltatomer i den første solvationsskal. Dette resulterer i en stærkt hæmmet rotationel bevægelse på grund af den asymmetriske potentialeenergioverflade for rotation. Den reducerede rotationelle bevægelse kobles til dens translationelle bevægelse og reducerer dens samlede diffusivitet sammenlignet med nitrationen.
- Sulfat-ionen (SO42-): Selvom sulfat er symmetrisk i sin ladningsfordeling (tetraedrisk geometri), udviser den på grund af sine højere partielle ladninger på de konstituerende atomer en meget langsom rotationel afslapning og translationel dynamik. Den stærke hydrogenbinding med de omgivende vandmolekyler gør rotationelle springbevægelser sjældnere sammenlignet med nitrat.
En modelberegning, hvor man gradvist indfører asymmetri i ladningsfordelingen af en hypotetisk ion med nitratlignende geometri, viser tydeligt, hvordan både rotationel afslapning og diffusivitet aftager med stigende asymmetri. Dette beviser, at for asymmetriske ioner bidrager reduceret rotationel bevægelse til en lavere diffusivitet.
Sammenligning af Sulfat, Ammonium og Metan
En sammenlignende undersøgelse af sulfat (SO42-), ammonium (NH4+) og metan (CH4) – hvor de to førstnævnte er ioner med tetraedrisk geometri, og sidstnævnte er et uladet molekyle med samme geometri – giver yderligere indsigt:
| Ion/Molekyle | DEksperimentel (x10-5 cm2/S) | DSimuleret (x10-5 cm2/S) | τ2 (ps) (Rotationel afslapningstid) |
|---|---|---|---|
| Sulfat (SO42-) | 1.065 | 0.95 | 11.66 |
| Ammonium (NH4+) | 1.957 | 2.15 | 8.39 |
| Metan (CH4) | ... | 3.1619 | 0.071 |
Tabelen viser, at metan, som et hydrofobt molekyle, har markant hurtigere translationel og rotationel dynamik. Sulfat har den langsomste dynamik på grund af dens høje partielle ladninger. Ammoniumioner er hurtigere end sulfat, men deres rotationelle dynamik er stadig langsommere end nitrationers, selvom deres translationelle diffusivitet er sammenlignelig.
Disse eksempler understreger, at ioners samlede ladning, individuelle partielle ladninger, molekylens symmetri og geometri alle spiller komplekse roller i at bestemme både strukturelle og dynamiske egenskaber for polyatomiske ioner.
Modekoblingsteori (MCT) for Polyatomiske Ioner
For at håndtere den øgede kompleksitet af polyatomiske ioner er Modekoblingsteori (MCT) blevet yderligere udviklet. I modsætning til monoatomiske ioner, hvor solventmolekylernes orienteringsafslapning er den primære koblingsmekanisme, skal man for polyatomiske solutter også inkludere soluttens egen orientering i kraft-kraft-tidskorrelationsfunktionen. Dette resulterer i komplekse udtryk, der kan redegøre for de indviklede koblinger.
MCT giver en dybdegående forståelse ved at sammenligne en "rotationelt frossen" (RF) ion (hvor rotation er forhindret) med en "rotationelt mobil" (RM) ion (der kan koble til vandets hydrogenbindingsnetværksdynamik). Diffusiviteten af rotationelt mobile ioner er typisk lavere end for rotationelt frosne, hvilket skyldes et skift i frekvens forårsaget af selvrotationen.
Teorien kan kvantitativt beskrive den rolle, som symmetri i ladningsfordelingen spiller for polyatomiske ioners rotationelle afslapning og translationelle diffusion. For eksempel har MCT-formalismen vist sig at stemme godt overens med simuleringsresultater for nitrationer og modelsystemer med varierende asymmetri i ladningsfordelingen, hvilket bekræfter, at stigende asymmetri fører til reduceret rotationel bevægelse og dermed lavere diffusivitet.
MCT's styrke ligger i dens evne til at inkorporere både enkeltpartikeldynamik og kollektiv dynamik, herunder lokalt strukturelle ændringer i solventet omkring ionerne. Dette gør den til et uundværligt værktøj til at opnå en detaljeret, mikroskopisk forståelse af ioners dynamik, som traditionelle hydrodynamiske modeller ikke kan fange.
Ofte Stillede Spørgsmål
Hvad får H+ og OH- til at være så mobile?
Den ekstremt høje mobilitet af protoner (H+) og hydroxidioner (OH-) i vand skyldes Grotthus-mekanismen. I stedet for at bevæge sig fysisk gennem opløsningen overføres den elektriske ladning via en serie af hurtige hydrogenbindingsskift mellem vandmolekyler. Dette betyder, at ladningen bevæger sig effektivt over lange afstande, selvom de individuelle atomer kun bevæger sig en kort strækning.
Hvorfor fejler kontinuummodeller for ioner?
Kontinuummodeller, som Stokes-Einstein, antager, at solventet er et homogent, viskøst medium, og at solutpartiklen er meget større end solventmolekylerne. For ioner i vand er disse antagelser ofte ugyldige. Ion-dipol-interaktionerne er langtrækkende og anisotropiske, ionernes størrelse er sammenlignelig med vandmolekylerne, og modellerne kan ikke fange de molekylære detaljer og den ultrahurtige dynamik af solventafslapning, som er afgørende for ioners bevægelse.
Hvordan påvirker ionstørrelse mobiliteten?
Ifølge simple hydrodynamiske modeller skulle mobiliteten falde med stigende ionstørrelse. Men for monoatomiske ioner ser man et "Walden-produktets sammenbrud", hvor mobiliteten ikke følger denne simple relation. Dette skyldes, at både Stokes' friktion (der stiger med størrelsen) og dielektrisk friktion (der falder for større ioner efter et vist punkt) spiller ind. For polyatomiske ioner er billedet endnu mere komplekst, da ionens egen rotation og ladningsfordeling også påvirker den effektive størrelse og interaktionen med solventet.
Hvad er betydningen af ultrahurtig solvation dynamik?
Ultrahurtig solvation dynamik refererer til den meget hurtige (inden for femtosekunder) afslapning af solventpolarisationen omkring en nyligt ladet eller exciteret ion. Denne hurtige afslapning mindsker den dielektriske friktion på en bevægende ion betydeligt. Hvis denne ultrahurtige komponent ikke tages i betragtning, overvurderer traditionelle modeller friktionen og undervurderer dermed mobiliteten.
Hvorfor er polyatomiske ioner mere komplekse at studere?
Polyatomiske ioner er mere komplekse på grund af deres fordelte ladninger, specifikke geometri og interne rotationelle frihedsgrader. Deres bevægelse er ikke kun bestemt af translationel friktion, men også af rotationel friktion og en stærk kobling mellem ionens egen translationelle og rotationelle bevægelse (translation-rotation kobling). Derudover kan deres asymmetriske ladningsfordeling og interaktioner med vandmolekylernes springbevægelser skabe yderligere kompleksiteter, der ikke ses i monoatomiske ioner.
Hvordan påvirker molekylær symmetri ionmobiliteten?
Molekylær symmetri i ladningsfordelingen har en signifikant indflydelse på polyatomiske ioners dynamik. Symmetriske ioner (som nitrat) har ofte hurtigere rotationel og translationel dynamik, fordi de kan gennemgå mere ubesværede rotationelle springbevægelser i solventet. Asymmetriske ioner (som acetat) oplever en hæmmet rotationel bevægelse på grund af en mere kompleks potentialeenergioverflade, hvilket igen reducerer deres translationelle diffusivitet. Dette skyldes en stærkere, mere retningsbestemt interaktion med solventmolekylerne.
Konklusion
Forståelsen af ionmobilitet og solvation dynamik er et komplekst, men afgørende felt inden for kemi og fysik. Fra de simple monoatomiske ioner til de indviklede polyatomiske arter har vi set, hvordan traditionelle hydrodynamiske modeller ofte kommer til kort, og hvorfor mikroskopiske tilgange som Modekoblingsteori (MCT) er uundværlige. Faktorer som ionens størrelse, ladningsfordeling, geometri, solventets viskositet og den ultrahurtige solvation dynamik spiller alle en rolle i at diktere ioners bevægelse.
Især for polyatomiske ioner afslører studiet af translation-rotation kobling og solvent-ion interaktioner en rigdom af dynamiske fænomener. Evnen til at forudsige og forklare disse bevægelser er ikke blot af akademisk interesse, men har vidtrækkende implikationer for områder som batteriteknologi, vandbehandling, biologiske processer og industriel kemi. Den fortsatte udvikling af teoretiske modeller og avancerede simuleringsmetoder vil utvivlsomt føre til en endnu dybere forståelse af disse fundamentale processer.
Hvis du vil læse andre artikler, der ligner Ionmobilitet: En Dybere Forståelse, kan du besøge kategorien Mobil.